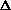 S = Q/T
S = Q/TEquation 2
| The Definitions of Entropy |
The popular literature is littered with articles, papers, books, and various & sundry other sources, filled to overflowing with prosaic explanations of entropy. But it should be remembered that entropy, an idea born from classical thermodynamics, is a quantitative entity, and not a qualitative one. That means that entropy is not something that is fundamentally intuitive, but something that is fundamentally defined via an equation, via mathematics applied to physics. Remember in your various travails, that entropy is what the equations define it to be. There is no such thing as an "entropy", without an equation that defines it.
Entropy was born as a state variable in classical thermodynamics. But the advent of statistical mechanics in the late 1800's created a new look for entropy. It did not take long for Claude Shannon to borrow the Boltzmann-Gibbs formulation of entropy, for use in his own work, inventing much of what we now call information theory. My goal here is to shwo how entropy works, in all of these cases, not as some fuzzy, ill-defined concept, but rather as a clearly defined, mathematical & physical quantity, with well understood applications.
Entropy and Classical Thermodynamics
Classical thermodynamics developed during the 19th century, its primary architects being Sadi Carnot, Rudolph Clausius, Benoit Claperyon, James Clerk Maxwell, and William Thomson (Lord Kelvin). But it was Clausius who first explicitly advanced the idea of entropy (On Different Forms of the Fundamental Equations of the Mechanical Theory of Heat, 1865; The Mechanical Theory of Heat, 1867). The concept was expanded upon by Maxwell (Theory of Heat, Longmans, Green & Co. 1888; Dover reprint, 2001). The specific definition, which comes from Clausius, is as shown in equation 1 below.
In equation 1, S is the entropy, Q is the heat content of the system, and T is the temperature of the system. At this time, the idea of a gas being made up of tiny molecules, and temperature representing their average kinetic energy, had not yet appeared. Carnot & Clausius thought of heat as a kind of fluid, a conserved quantity that moved from one system to the other. It was Thomson who seems to have been the first to explicity recognize that this could not be the case, because it was inconsistent with the manner in which mechanical work could be converted into heat. Later in the 19th century, the molecular theory became predominant, mostly due to Maxwell, Thomson and Ludwig Boltzmann, but we will cover that story later. Suffice for now to point out that what they called heat content, we would now more commonly call the internal heat energy.
The temperature of the system is an explicit part of this classical definition of entropy, and a system can only have "a" temperature (as opposed to several simultaneous temperatures) if it is in thermodynamic equilibrium. So, entropy in classical thermodynamics is defined only for systems which are in thermodynamic equilibrium.
As long as the temperature is therefore a constant, it's a simple enough exercise to differentiate equation 1, and arrive at equation 2.
S = Q/T
Here the symbol "" is a representation of a finite increment, so
that S indicates a "change" or "increment" in
S, as in S = S1 - S2, where S1
and S2 are the entropies of two different equilibrium states,
and likewise Q. If Q
is positive, then so is S, so if the internal heat energy
goes up, while the temperature remains fixed, then the entropy S goes up.
And, if the internal heat energy Q goes down (Q
is a negative number), then the entropy will go down too.
Clausius and the others, especially Carnot, were much interested in the ability to
convert mechanical work into heat energy, and vice versa. This idea can lead us to
an alternate form for equation 2, that will be useful later on. Suppose you pump
energy, U, into a system, what happens? Part of the energy
goes into the internal heat content, Q, making Q a
positive quantity, but not all of it. Some of that energy could easily be expressed
as an amount of mechanical work done by the
system (W, such as a hot gas pushing against a piston in
a car engine). So that
Q = U - W, where
U is the energy input to the system, and W
is the part of that energy that goes into doing work. The difference between them is the amount
of energy that does not participate in the work, and goes into the heat resevoir as
Q. So a simple substitution allows equation 2 to be re-written
as equation 3.
S = (U - W)/T
This alternate form of the equation works for heat taken out of a system (U
is negative) or work done on a system (W is negative), just as well. So
now we have a better idea of the classical relation between work, energy and entropy. Before we
go on to the more advanced topic of statistical mechanics, we will take a useful moment to apply
this to classical chemistry.
Entropy and Physical Chemistry
At the same time that engineers & physicists were laying the foundations for thermodynamics, the chemists were not being left out. Classical entropy plays a role in chemical reactions, and that role is exemplified in equation 4 below.
S = (H - F)/T
Of course, this looks just like equation 3 with different letters, and so it is. Here, we are
not much interested in the physicists approach of describing the state of a "static" system,
as does equation 1. The real interest for the chemist, is to predict whether or not a given
chemical reaction will go. In equation 4, H is the enthalpy, and F is
the free energy (also known as the Gibb's free energy). Likewise, H
and F are incremental variations of those quantities, and S
is an incremental change in the entropy of the chemical system, in the event of a chemical reaction.
A little algebra, leading to equation 5, will maybe make things just a little easier to see.
F = H - TS
The significance of this equation is that it is the value of F which
tells you whether any give chemical reaction will go forward spontaneously, or whether it needs
to be pumped. The enthalpy, H, is the heat content of the system, and so the change in
enthalpy, H, is the change in heat content of the system. If that
value is smaller than TS, then F will
be negative, and the reaction will proceed spontaneously; the TS
term represents the ability to do the work required to make the reaction happen. However,
if F is positive, such that H is
greater than TS, then the reaction will not happen spontaneously;
we still need at least F worth of energy to make it happen.
Note that a positive free energy does not mean that the reaction will not happen, only that
it will not happen spontaneously in the given environment. It can still be pushed
or pumped into happening by adding energy, or setting the reaction in a higher temperature
environment, making T larger as well as TS, and perhaps
driving it far enough to make F negative.
Entropy and Statistical Mechanics
In the later 1800's, Maxwell, Ludwig Boltzmann and Josiah Willard Gibbs extended the ideas of classical thermodynamics, through the new "molecular theory" of gases, into the domain we now call statistical mechanics. In classical thermodynamics, we deal with single extensive systems, whereas in statistical mechanics we recognize the role of the tiny constituents of the system. The temperature, for instance, of a system defines a macrostate, whereas the kinetic energy of each molecule in the system defines a microstate. The macrostate variable, temperature, is recognized as an expression of the average of the microstate variables, an average kinetic energy for the system. Hence, if the molecules of a gas move faster, they have more kinetic energy, and the temperature naturally goes up.
Equation 6 below is the general form of the definition of entropy in statistical mechanics, as first derived by Boltzmann. You can see Boltzmann's own derivation in his Lectures on Gas Theory (available as a Dover reprint), but more modern treatments might be easier to follow, such as Statistical Physics by Gregory H. Wannier, or The Principles of Statistical Mechanics by Richard C. Tolman (both also available as Dover reprints).
 [Pilog(Pi)]
[Pilog(Pi)]
In this equation, Pi is the probability that particle "i" will
be in a given microstate, and all of the Pi are evaluated for the same
macrostate of the system. The symbol (an upper case Greek sigma)
is a mathematical instruction to add up everything to the right of it. In this case, it
means to add up the product of Pi times log(Pi) for
all of the "i" particles. The "k" out in front is an arbitrary constant,
which determines the units of measure of entropy, and in thermodynamics is Boltzmann's constant
(1.380658×10-23 Joules/Kelvin), but its value could just as easily be arbitrarily
set to 1 without affecting the generality of the arguments presented here. The negative
sign is there because the probability is a number between 0 and 1, so its logarithm will
always be negative, so the negative sign out front cancels the negative sign induced by
taking the log of a number less than 1.
Contrary to the definition seen in equation 1, neither the temperature nor the heat energy appear explicitly in this equation. However, the restriction that all of the microstate probabilities must be calculated for the same macrostate, assures that, as in the earlier case, the system must be in a state of thermal equlibrium.
Equation 6 treats the microstate probabilities individually. However, if all of the probabilities are the same, then we can simplify equation 6 to equation 7.
In this simplified form, the only thing we have to worry about is "N", which is the total number of microstates available to the system. Be careful to note that this is not the total number of particles, but rather the total number of microstates that the particles could occupy, with the constraint that all such microstate collections would show the same macrostate.
I have separated quantum mechanics from statistical mechanics, to avoid confusion, and to avoid the implication that something important may have been overlooked. However, since the two both deal intimately with statistics & probabilities, it should come as no surprise that entropy is handled by the two disciplines in much the same way. The quantum mechanical definition of entropy is identical to that given for statistical mechanics, in equation 6. The only real difference is in how the probabilities are calculated. Quantum mechanics has its own, peculiar rules for doing that, but they are not relevant to the fundamental definition of entropy. As in the previous cases, the Pi are microstate probabilites, and they must all be calculated for the same macrostate.
Entropy and Information Theory
The work done, primarily by Boltzmann & Gibbs, on the foundations of statistical mechanics, is of profound significance that can hardly be overestimated. In the hands of Clausius and his contemporarys, entropy was an important, but strictly thermodynamic property. Outside of physics, it simply had no meaning. But the mathematical foundations of statistical mechanics are applicable to any statistical system, regardless of its status as a thermodynamic system. So it is by the road of statistical mechanics, that we are able to talk about entropy in fields outside of thermodynamics, and even outside of physics per se.
Perhaps the first major excursion of entropy into new domains, comes at the hands of Claude Shannon, widely recognized as the father of modern communication & information theory (his classical 1948 paper A Mathematical Theory of Communication is on the web).
[Pilog(Pi)]If this looks familiar, it's not an accident. It's quite the same as equation 6 above, the definition of entropy in statistical mechanics. In A Mathematical Theory of Communication, appendix 2, Shannon proves his Theorem 2, that this Boltzmann entropy is the only function which satisfy's the requirements for a function to measure the uncertainty in a message (where a "message" is a string of binary bits). In this case, the constant k is recognized as only setting the units; it is arbitrary, and can be set equal to exactly 1 without any loss of generality (see the discussion in Shannon's paper, begining with section 6 "Choice, uncertainty and entropy"). In this case the probabilty Pi is the probability for the value of a given bit (usually a binary bit, but not necessarily).
In Shannon information theory, the entropy is a measure of the uncertainty over the true content of a message, but the task is complicated by the fact that successive bits in a string are not random, and therefore not mutually independent, in a real message. Also note that "information" is not a subjective quantity here, but rather an objective quantity, measured in bits.
So far, we have looked at entropy in its most common, and well known forms. Most ordinary applications will use one of these entropies. But, there are other forms of entropy beyond what I have shown. For instance, Brazilian Mathematician Constantino Tsallis has derived a generalized form for entropy, which reduces to the Boltzmann-Gibbs entropy in our equation 6, as a special case, but can also be used to describe the entropy of a system for which our equation 6 would not work (see "Justifying the Tsallis Formalism"). Hungarian mathematician Alfréd Rényi was able to construct the proper entropy for fractal geometries (see "The world according to Rényi: thermodynamics of fractal systems""). There are others besides these, but the entropies of Tsallis & Rényi are the ones that seem to be under the most active current consideration.
These generalized forms of entropy, of relatively recent origin, serve to show that "entropy" is not just an old friend that we know quite well, as in classical thermodynamics, but also a concept that is rich in new ideas & scientific directions.
Is Entropy a Measure of "Disorder"?
Let us dispense with at least one popular myth: "Entropy is disorder" is a common enough assertion, but commonality does not make it right. Entropy is not "disorder", although the two can be related to one another. For a good lesson on the traps and pitfalls of trying to assert what entropy is, see Insight into entropy by Daniel F. Styer, American Journal of Physics 68(12): 1090-1096 (December 2000). Styer uses liquid crystals to illustrate examples of increased entropy accompanying increased "order", quite impossible in the entropy is disorder worldview. And also keep in mind that "order" is a subjective term, and as such it is subject to the whims of interpretation. This too mitigates against the idea that entropy and "disorder" are always the same, a fact well illustrated by Canadian physicist Doug Craigen, in his online essay "Entropy, God and Evolution".
The easiest answer to the question, "What is entropy?", is to reiterate something I said in the introduction: Entropy is what the equations define it to be. You can interpret those equations to come up with a prosey explanation, but remember that the prose & the equations have to match up, because the equations give a firm, mathematical definition for entropy, that just won't go away. In classical thermodynamics, the entropy of a system is the ratio of heat content to temperature (equation 1), and the change in entropy represents the amount of energy input to the system which does not participate in mechanical work done by the system (equation 3). In statistical mechanics, the interpretation is more general perhaps, where the entropy becomes a function of statistical probability. In that case the entropy is a measure of the probability for a givem macrostate, so that a high entropy indicates a high probability state, and a low entropy indicates a low probability state (equation 6).
Entropy is also sometimes confused with complexity, the idea being that a more complex system must have a higher entropy. In fact, that is in all liklihood the opposite of reality. A system in a highly complex state is probably far from equilibrium and in a low entropy (improbable) state, where the equilibrium state would be simpler, less complex, and higher entropy.
Move on to the second law of thermodynamics
Go back to The Collected Writings of Tim Thompson
Go all the way to Tim Thompson's Home Page
Page dated 19 February 2002